3D structure prediction based on amino acid sequnces
What is I-TASSER server?
|
I-TASSER
server is an internet service for protein structure and function predictions.
It allows acedemic users to automatically generate high-quality predictions of
3D structure and biological function of protein molecules from their amino acid
sequences.
How does I-TASSER generate
structure and function predictions?
|
When
users submit an amino acid sequence, the server first tries to retrieve
template proteins of similar folds (or super-secondary structures) from the PDB
library by LOMETS, a locally installed meta-threading
approach.
In
the second step, the continuous fragments excised from the PDB templates are
reassembled into full-length models by replica-exchange Monte Carlo simulations
with the threading unaligned regions (mainly loops) built by ab initio
modeling. In cases where no appropriate template is identified by LOMETS,
I-TASSER will build the whole structures by ab initio modeling. The low
free-energy states are identified by SPICKER through
clustering the simulation decoys.
In
the third step, the fragment assembly simulation is performed again starting
from the SPICKER cluster centroids, where the spatial restrains collected from
both the LOMETS templates and the PDB structures by TM-align are
used to guide the simulations. The purpose of the second iteration is to remove
the steric clash as well as to refine the global topology of the cluster
centroids. The decoys generated in the second simulations are then clustered
and the lowest energy structures are selected. The final full-atomic models are
obtained by REMO which builds the atomic details from the selected I-TASSER
decoys through the optimization of the hydrogen-bonding network (see Figure 1).
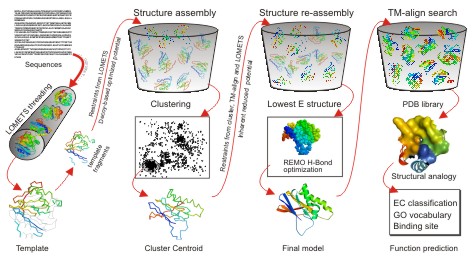
Figure 1. I-TASSER protocol for protein structure and function prediction.
For
predicting the biological function of the protein (the last column at Figure
1), the I-TASSER server matches the predicted 3D models to the proteins in 3
independent libraries which consist of proteins of known enzyme classification
(EC) number, gene ontology (GO) vocabulary, and ligand-binding sites. The final
results of function predictions are deduced from the consensus of top
structural matches with the function scores calculated based on the confidence
score of the I-TASSER structural models, the structural similarity between
model and templates as evaluated by TM-score,
and the sequence identity in the structurally aligned regions [A similar
approach to structure-based function annotation was proposed by Brylinski and
Skolnick (PNAS 2008. 205:129) who tried to match the target structures on the threading
templates. Here the I-TASSER server matches the target models on all template
proteins in the libraries].
What are the performances of
I-TASSER server compared with other methods?
|
CASP (or
Critical Assessment of Techniques for Protein Structure Prediction) is
a community-wide experiment for testing the state-of-the-art of protein
structure predictions which takes place every two years since 1994. The
experiment (often referred as a competition) is strictly blind because the
structures of testing proteins are unknown to the predictors.
The
I-TASSER server (as "Zhang-Server") participated in the Server
Section of 7th (2006), 8th (2008),
and 9th CASPs (2010), and was ranked as the No
1 server in CASP7 and CASP8. In CASP9, I-TASSER server and QUARK (another
server from our lab) were ranked as No 1 and No 2 servers, respectively. The
detailed rank results can be seen here for CASP7, CASP8,
and CASP9. Figure 2 shows histograms of the Z-score
of GDT-TS scores of all servers in CASP7 (68 servers), CASP8 (81 servers), and
CASP9 (81 servers).

Figure 2. Histogram of Z-scores of all server groups at CASP7, CASP8 and CASP9.
What are the output of the
I-TASSER server if you submit a seqeunce?
|
The
output of the I-TASSER server include:
·
Up to five full-length
atomic models (ranked based on cluster density)
·
Estimated accuracy of
the predicted models (including a confidence score of all models, and predicted
TM-score and RMSD for the first model)
·
GIF images of the
predicted models
·
Predicted secondary
structures
·
Predicted solvent
accessibility
·
Top 10 threading alignment
from LOMETS
·
Top 10 proteins in PDB
which are structurally closest to the predicted models
·
Predicted Enzyme
Classification and the confidence score
·
Predicted GO terms and
the confidence score
·
Predicted
ligand-binding sites and the confidence score
·
An image of the
predicted ligand-binding sites
How to use known information (e.g.
templates and function) to improve I-TASSER modeling?
|
If
users know some information about the structure of the modeled proteins, the
information can be conveniently uploaded to the I-TASSER server. These
information can significantly improve the quality of structural and function
predictions.
The
I-TASSER server currently accepts two types of user-specified restraints:
(1) inter-residue contant and distance
restraints;
(2) template structures and template-target alignments.
(2) template structures and template-target alignments.
The
server provides 4 convenient options to assign the restraints:
·
Assign contact/distance
restraints: If you know what atom pairs should be in contact or in some
distances, you can use this option to upload a text file including the contact
and/or distance information of atom pairs.
·
Specify template
without alignment: If you want I-TASSER to use a specific PDB structure as a
template, you can use this option specify the PDB structure. You only need to
type in the PDBID:ChainID, e.g. 1wor:A without specifying the target-template
alignments. If the chain information is not present in the PDB file, indicate
the ChainID using "_". I-TASSER will first fetch the structure from
the PDB library and then generate the target-template alignment based on our
in-house alignment tool, MUSTER.
·
Specify template
without alignment: You can actually use any 3D structure as the template, which
does not necessary exist in the PDB library. In this case, you can use this
option to upload the 3D structure. This structure file must be in the standard
PDB format. You do not need to input the target-template alignments. I-TASSER
will generate target-template alignment based on our in-house alignment tool,
MUSTER.
·
Specify template with
alignment: This option allows you (usually the advanced users) to specify both
template structure and the target-template alignment.
Can I exclude some proteins from
the I-TASSER template library?
|
I-TASSER
needs templates to generate high-resolution structure predictions. In general,
excluding close templates will decrease the quality of the I-TASSER modeling.
However, users can exclude some templates from the I-TASSER template library
for some special purposes (e.g. knowning some templates are different from
target, or benchmark testing of the current algorithms).
The
I-TASSER server accept two ways of template excludings:
·
Exclude
templates that are homologous to the query protein: The users can use this option to exclude
templates from the I-TASSER template library, which are homologous to the query
protein. The homology is defined based on the sequence identity cutoff, i.e.
the number of identical residue between template and query divided by the total
number of residues in the query sequence. For example, if you type
"60%", I-TASSER will automatically exclude all templates which have a
sequence identity >60% to the query protein. The minimum cutoff is set at
25% and all value below 25% will return as 25%.
·
Exclude
specific template proteins: This
option allows users to upload a list of template structures that will be
excluded from the I-TASSER template library. As the PDB library is redundant
and same protein can exist as multiple entries, I-TASSER server will by default
exclude the user-specified templates as well as all templates that have a
sequence identity >90% to the specified templates. Users can also specify a
different sequence identity cutoff, e.g. 70%, where I-TASSER will exclude all
templates with a sequence identity >70% to specified template proteins.
The format of the file should be
"PDBID:ChainID %Sequence_Identity", e.g.
1wor:A 70
3mxu:A 80
1zko:B 40
3mxu:A 80
1zko:B 40
OR
1wor:A
3mxu:A
1zko:B
3mxu:A
1zko:B
What is new?
|
·
2011/10/15: Visualization was enabled for the top 10
proteins analogous to the I-TASSER models and for the enzyme commission
predictions.
·
2011/10/09: The function homology search was extended to
the entire PDB library. This helps increase the coverage and accuracy of the
structure-based function of the I-TASSER server.
·
2011/08/01: The I-TASSER server had the 20000th user
registered (Congratulations!).
·
2011/03/28: The first version of the I-TASSER Suite
(Version 1.1) was publicly released for download and installation. It is free
for academic and non-profit users.
How long does it take for I-TASSER
to generate the predictions for your protein?
|
It
usually takes server hours to 1~2 days from submitting a sequence to receiving
the prediction results. But if too many sequences are accumulated in the queue,
the procedure may take a much longer time. The time also depends on the protein
size and a smaller protein takes short time than a larger protein.
Currently,
the major time consuming part in the I-TASSER protocol is the structural
refinement assembly simulations. For those users who want a quicker reponse or
those who do not need a refined models, we recommend them to use our LOMETS
(meta-server) or MUSTER
(single-server fold-recognition). Because these two server do not
attempt to refine the threading models, the response time is faster than the
I-TASSER server.
No comments:
Post a Comment